
Τι είναι η Νόσος von Hippel-Lindau (VHL)
 Η νόσος von Hippel-Lindau (VHL) είναι μια σπάνια αυτοσωματική επικρατούσα νόσος με επιπολασμό 1:36.000 γεννήσεις. Κλινικά, σχετίζεται με την ανάπτυξη καλοήθων και κακοήθων όγκων σε πολλά όργανα. Συνδέεται με μεταλλάξεις του ογκοκατασταλτικού γονιδίου VHL (χρωμόσωμα 3). Η αναλογία στα δύο φύλλα, λόγω του αυτοσωμικού χαρακτήρα της νόσου, είναι 1:1. Το ποσοστό των ασθενών που πάσχουν από την κληρονομική μορφή της νόσου ανέρχεται στο 80%, με το υπόλοιπο 20% να μην έχει οικογενειακό ιστορικό και να προέρχεται από νέες μεταλλάξεις (de-novo) των υπεύθυνων γονιδίων.
Η νόσος von Hippel-Lindau (VHL) είναι μια σπάνια αυτοσωματική επικρατούσα νόσος με επιπολασμό 1:36.000 γεννήσεις. Κλινικά, σχετίζεται με την ανάπτυξη καλοήθων και κακοήθων όγκων σε πολλά όργανα. Συνδέεται με μεταλλάξεις του ογκοκατασταλτικού γονιδίου VHL (χρωμόσωμα 3). Η αναλογία στα δύο φύλλα, λόγω του αυτοσωμικού χαρακτήρα της νόσου, είναι 1:1. Το ποσοστό των ασθενών που πάσχουν από την κληρονομική μορφή της νόσου ανέρχεται στο 80%, με το υπόλοιπο 20% να μην έχει οικογενειακό ιστορικό και να προέρχεται από νέες μεταλλάξεις (de-novo) των υπεύθυνων γονιδίων.
Κλινική Εικόνα
Κλινικά η νόσος von Hippel-Lindau χαρακτηρίζεται από αιμαγγειώματα αμφιβληστροειδούς (30% – 60% των ασθενών), διαυγοκυτταρικά καρκινώματα νεφρού (clear cell Renal Cell Carcinoma-RCC στο 30% – 50% των ασθενών), αιμαγγειοβλαστώματα της παρεγκεφαλίδας και του νωτιαίου μυελού (60% – 80% των ασθενών), φαιοχρωμοκυττώματα (7% – 20% των ασθενών), και λιγότερο συχνά κύστεις παγκρέατος και νευροενδοκρινικούς όγκους.
Για την ιστορία αναφέρουμε ότι ο Engen Von Hippel, περιέγραψε για πρώτη φορά αυτού του τύπου τα αγγειώματα στο μάτι. Ο Dr.Arvid Lindau, ήταν Σουηδός παθολόγος που περιέγραψε αναλυτικά τα αιμαγγειώματα σε παρεγκεφαλίδα και σπονδυλική στήλη το 1926.
Ταξινόμηση των Ασθενών με Νόσο VHL
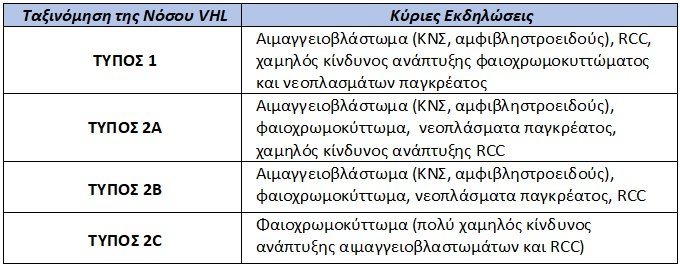
Αρχικά οι ασθενείς με νόσο von Hippel-Lindau (VHL) κατηγοριοποιούνται σε δύο ομάδες. Η ομάδα 1 περιλαμβάνει ασθενείς χαμηλού κινδύνου ανάπτυξης φαιοχρωμοκυττώματος, ενώ η ομάδα 2 τους ασθενείς υψηλού κινδύνου για ανάπτυξη φαιοχρωμοκυττώματος. Η ομάδα 2 υποδιαιρείται με την σειρά της σε τρεις υπότυπους: τον τύπο 2A (ασθενείς με φαιοχρωμοκύττωμα, αιμαγγειοβλαστώματα του κεντρικού νευρικού συστήματος ή/και αιμαγγειώματα αμφιβληστροειδούς), τον τύπο 2B (ασθενείς 2Α που εμφανίζουν επιπλέον διαυγοκυτταρικό καρκίνωμα νεφρού ή/και κύστεις παγκρέατος), τον τύπο 2C (περιλαμβάνει ασθενείς μόνο με φαιοχρωμοκύττωμα).
Διάγνωση
Η διάγνωση της νόσου von Hippel-Lindau (VHL) σε ασθενείς χωρίς οικογενειακό ιστορικό θα πρέπει να γίνεται όταν συνυπάρχουν πολλαπλοί όγκοι όπως αιμαγγειοβλαστώματα του αμφιβληστροειδούς ή/και του κεντρικού νευρικού συστήματος, ή ένα μόνο αιμαγγειοβλάστωμα συν ένα από τα άλλα χαρακτηριστικά της νόσου (βλ. πίνακα), ενώ σε ασθενείς με οικογενειακό ιστορικό αρκεί η παρουσία ενός και μόνο αιμαγγειοβλαστώματος του κεντρικού νευρικού συστήματος, ή ενός και μόνο φαιοχρωμοκυττώματος ή διαυγοκυτταρικού καρκινώματος νεφρού. Η γενετική μοριακή διάγνωση της νόσου έχει διευκολύνει σε μεγάλο βαθμό την αξιολόγηση και τη διαχείριση των οικογενειών, μέλη των οποίων έχουν την ασθένεια. Η ικανότητα ανίχνευσης των μεταλλάξεων ανέρχεται σχεδόν στο 100%.
Οι κύστεις του νεφρού συνήθως, αλλά όχι πάντα, προηγούνται της ανάπτυξης των νεφρικών όγκων. Η νόσος von Hippel-Lindau σχετίζεται με RCC τα οποία και εκδηλώνονται νωρίς στη ζωή, με μέση ηλικία κατά τη διάγνωση τα 35 έτη. Η αθροιστική πιθανότητα ανάπτυξης RCC αυξάνεται γεωμετρικά φθάνοντας το 70% στην ηλικία των 60 ετών. Οι μεταστάσεις των RCC αποτελούν το κύριο αίτιο θανάτου αυτών των ασθενών. Ο καλύτερος τρόπος διαχείρισης αυτών των όγκων είναι η παρακολούθηση τους έως ότου φθάσουν τα 3 cm σε μέγεθος. Πέραν των 3 cm, ο κίνδυνος μετάστασης αυξάνεται και η χειρουργική επέμβαση-εκτομή κρίνεται απαραίτητη. Για αυτό το λόγο οι ασθενείς υψηλού κινδύνου θα πρέπει να υποβάλλονται ετησίως σε παρακλινικό έλεγχο (βυθοσκόπηση, MRI εγκεφάλου, CT ή U/S κοιλιάς). Ετήσιος, θα πρέπει να είναι και ο προσδιορισμός των επιπέδων των κατεχολαμινών/μετανεφρινών στο αίμα και στα ούρα.